有機合成化学、量子化学計算、時間分解レーザー分光を基盤として、高機能性フォトクロミック分子や有機ラジカルの開発を目指した物理有機化学の研究を行っています。
機能物質化学研究室
基礎解説Accounts
基礎解説 ⇒ T型フォトクロミック分子 HABI 高速フォトクロミック分子 ビラジカル
論文概要 ⇒ HABI 高速フォトクロミック分子 ビラジカル
T型フォトクロミック分子
- 自然界に見られるフォトクロミック分子
自然界ではフォトクロミック分子は様々な機能の発現を調節する光スイッチとして重要な役割を担っている。脊椎動物の網膜には、桿体、錐体とよばれる二種類の視細胞があり、前者は薄明視(明暗)を、後者は昼間視(色)を司っている。視細胞の光受容部には特有な視物質が含まれているが、最もよく研究されているのはロドプシンである[1]。ロドプシンは色素タンパク質で、その発色団は11-シスレチナールであり、タンパク質部分はオプシンとよばれている。11-シスレチナールは可視光を吸収するとall-トランスレチナールに光異性化し、オプシンから解離して、オプシンの構造変化を引き起こす。この光化学反応が視覚の初期過程である。
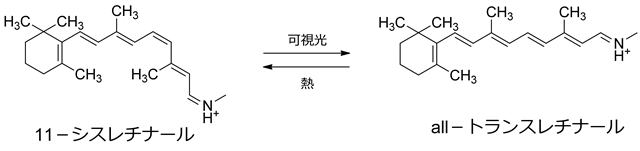
レチナールのシス−トランス光異性化反応
また、フィトクロムは植物の光形態形成を司っているフォトクロミック色素タンパク質である[2]。暗所で育った単子葉植物から抽出・精製したフィトクロム溶液では664nmに吸収極大を持つ赤色光吸収型フィトクロム(Pr)として存在するが、赤色光を吸収すると724nmに吸収極大を持つ近赤外光吸収型フィトクロム(Pfr)に変換する。一方、Pfrは近赤外光を吸収するとPrに変換する。このPrとPfrの間に起こる光変換は、繰り返し可逆的に起こすことができる。植物の種子に赤色光を当てた場合にはよく発芽するが、近赤外光を当てた場合にはほとんど発芽しないことから、Pfrの働きで発芽に関わる酵素の合成が誘導され、発芽が起きると考えられている。
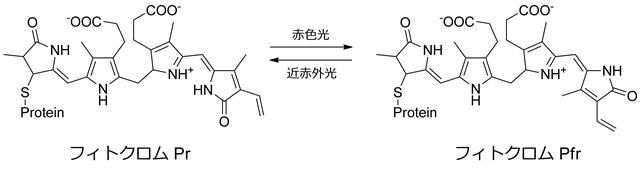
フィトクロムの光異性化反応
バクテリオロドプシン(bR)は古細菌(Archae)に属する高度好塩菌Halobacterium salinarumの細胞膜に存在する膜タンパク質で、ロドプシンと同じく発色団としてレチナールを持つ[3-6]。単量体の分子量は約26,000(248残基)で、それが三量体を単位に6方晶系の二次元結晶として、紫膜とよばれるパッチを形成している。bR中のレチナールはLys216とシッフ塩基結合(−CH=N−)を形成しているが、シッフ塩基はプロトン化(−CH=NH+−)している。bRが光を吸収すると、all-トランスレチナールは13-シスレチナールへ光異性化、プロトン移動、細胞外へプロトン放出、シッフ塩基の再プロトン化を経て、再度、最初のall-トランスレチナールへ戻ることで、細胞質側から細胞外側へ一つのプロトンを能動輸送する[7-10]。好塩菌はプロトン輸送によって生まれた電気化学的ポテンシャル差を利用してATP合成、鞭毛の回転、イオンの輸送などを行う。特筆すべきことに、bRのプロトン輸送過程はサイクル反応であり、野生型bRの光反応サイクルは約20ミリ秒程度である。これは、まさに自然界に存在する高速熱消色反応である。bRはフォトクロミズムに伴う分子構造変化を巧みに利用して光エネルギー変換を行っているが、最近では、高速原子間力顕微鏡を用いて、光励起に伴う変異体bRの実時間動態観察が報告されている[11,12]。
また、緑藻類の一種のクラミドモナスの眼点に分布するチャネルロドプシン(ChR)は可視光に応答して陽イオンを透過させ、膜電位を制御することにより走光性を制御している。ChRを遺伝子工学的にニューロンに導入・発現させることで、標的とするニューロンを光刺激により興奮または抑制することができる。光感受性タンパク質であるChRを用いた神経科学分野における光遺伝学(オプトジェネティックス)の研究は飛躍的に発展している。特筆すべきことに、bRやChRのイオン輸送過程はサイクル反応であり、bRの光反応サイクルは約20ミリ秒程度である。このように、ロドプシンファミリータンパク質はまさに光駆動型の高速イオンポンプであり、フォトクロミズムに伴う可逆的な高速分子構造変化を巧みに利用して光エネルギー変換を行っている。
室温程度の熱エネルギーで異性体Bから異性体Aに戻るT型フォトクロミック分子は光照射のON、OFFだけで物質の透過率、屈折率、反射率[13]、蛍光強度などをスイッチすることができるので、様々な用途に利用することができる。産業的に利用されているものとしては調光レンズが有名である。屋外の太陽光が強い環境では可視光を吸収する異性体Bに光異性化して可視光の透過率を下げることで、太陽光の眩しさを軽減し目の健康を守る眼鏡用レンズである。紫外線量の少ない屋内では、異性体Aに戻ることで速やかに退色して無色に近い状態になる。現在、製品として販売されている調光レンズでは、完全消色には数分から数十分の時間を要するが、熱消色反応のさらなる高速化に向けた研究が行われている。一方、T型フォトクロミック分子の熱消色反応速度をミリ秒からナノ秒程度にまで高速化することにより、その応用範囲は格段に広がる。これまでは専らフォトクロミズムの分子構造変化や色変化を利用した光応答システムが研究されてきたが、ロドプシンファミリータンパク質が行っているような光駆動分子ポンプを実現することができれば、これまでにない新しいタイプの光エネルギー変換システムの構築が可能になる。さらに、全光型論理回路[14-16]、超解像蛍光顕微鏡[17-29]、実時間光情報処理[30-33]などへの応用も期待されている。
- ベンゾピラン誘導体
代表的フォトクロミック分子の一つであるスピロピランは二つの異なるヘテロ環ユニットから構成されている。一つはベンゾピランユニットで、他方はインドリン環ユニットであるが、これら二つのユニットはスピロ炭素原子を共有して結合されており、互いに直交した立体配置をとっている。1966年、Beckerらはスピロピランの研究を進めている過程で、下図に示すベンゾピラン(2H-クロメン)がフォトクロミズムを示すことを見いだした[34,35]。無色のベンゾピランに紫外光を照射すると、スピロピランのフォトクロミズムと同様にC-O結合が切れて開環体が生成することで赤色に発色し、熱あるいは可視光照射で閉環体に戻る。スピロピラン類の発色体は双性イオン構造をとるのに対して、ベンゾピラン類の発色体はカルボニル基をもつ中性構造をとることが特徴である。
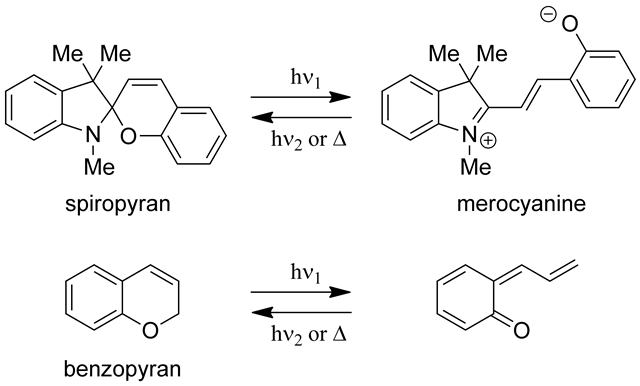
スピロピランとベンゾピランのフォトクロミズム
ベンゾピラン誘導体であるナフトピラン類は熱消色反応速度が比較的速く、繰り返し耐久性も高いために調光レンズへの応用に向けた研究が行われている[36-43]。ただ、研究草創期には発色体に関する詳細な記述は行われなかった。Beckerは米国特許に3,3-ジフェニル-3H-ナフト[2,1-b]ピランのフォトクロミック挙動について、「−40℃以下で紫外光を照射すると、オレンジ色に発色するが、室温では消色して無色になる」とだけ記している[44]。1986年にLenobleとBeckerは室温でのレーザーフラッシュフォトリシス測定を行い、開環体は430nmに吸収極大を持ち黄色を呈することを報告したが、発色体の半減期については明確に言及せず、400マイクロ秒まで安定であると記したに過ぎない[45]。一方、2H-ナフト[1,2-b]ピラン誘導体はプラスチック調光レンズとして実用化されており、代表的なものとしてPPG Industries社とEssilor International社の合弁会社であるTransitions Optical社が上市したTransitionsプラスチックレンズがある。
ナフトピラン誘導体
ナフトピラン類に紫外光を照射すると、発色体として二種類の異性体transoid-cis(TC)体とtransoid-trans(TT)体、および無色のAL(allenylnaphthol)体を生じる[46-48]。AL体は熱的に不安定であり低温条件下でのみ観測される。光反応の主生成物であるTC体は熱反応により速やかに元の閉環構造を持つ消色体に戻るが、TT体は熱的に安定であり長寿命であるため、熱消色反応過程はこれらに対応する速い減衰成分と遅い減衰成分の二成分を含む。もちろん置換基に依存することは言うまでもないが、TC体は数分から数十分で閉環体に戻るのに対して、TT体が完全に消失するまでには暗中で数十分から数時間を要する。すなわち、光定常状態に達した後に紫外光照射をやめると、TC体が比較的速やかに閉環体に戻ることで発色濃度は20%程度にまで退色するが、熱的に安定なTT体が残存するためにうっすらと発色した状態が長く続くことになる[38,48,49]。TT体はTC体を経由して閉環体に戻るが、可視光照射によりtrans-cis光異性化が起こり、TC体に異性化することで熱消色反応が加速される。したがって、紫外光照射を止めると同時に可視光を連続照射した場合、TT体はTC体に光異性化するために、暗中よりも速やかに退色する[48]。Delbaereらは高分解能19F NMRを用いてフルオロフェニル基を導入したナフトピラン誘導体の開環体の光化学および熱反応に関する詳細な速度論的研究を報告している[46]。
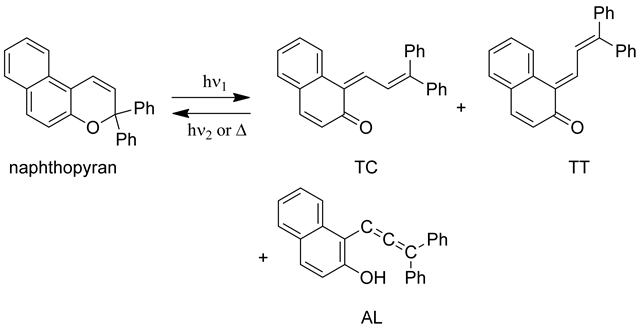
ナフトピランの光生成種
熱消色型フォトクロミック分子を調光材料に応用する場合、発色反応の量子収率、発色体の色調、熱消色反応速度、耐光性が重要になるが、ナフトピラン類のフォトクロミック特性は分子構造に大きく依存することから様々な分子設計が検討されてきた。特に、ナフトピランの5,6位にインデノ基を縮環させたインデノ縮合ナフトピランでは開環体のπ共役が広がることで、モル吸光係数の増大を伴う吸収極大波長の長波長シフトと、TC体の立体的不安定性増大に起因する熱消色反応の大幅な高速化が達成された[47,49,50]。このような分子設計戦略により発色状態の色調のバリエーションの広がりも含めて、調光レンズとしての性能を大幅に改善することが可能になり、国内外で数多くの特許が出願されている。さらに、sp3炭素に置換している二つのフェニル基を硫黄で架橋したインデノ縮合スピロ[チオキサンテン-ナフトピラン]類では発色体の半減期は室温トルエン中で40秒程度にまで高速化されるとともに、TT体に由来する長寿命成分も減少することが見いだされた[51]。他にもナフトピランの5,6位にジメチルチオフェン環を縮環させたチエノ-2H-クロメンが報告されており、TC体の室温での半減期は325ミリ秒と大幅な高速化が達成されているが、濃度が0.1ミリモルのトルエン溶液に室温で40Wm−2の紫外光を連続照射しても発色を目視することはできなかった[52]。これは、熱消色反応が速すぎて、光定常状態で十分な量の発色体が蓄積しなかったことに起因する。一方で、インデノ縮合ナフトピランの発色体でも熱的に安定なTT体が生成するために、さらなる改良が求められている。
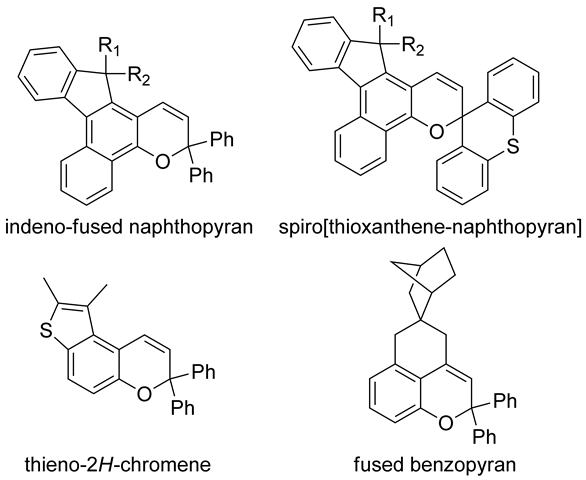
熱消色反応の高速化を実現したナフトピラン誘導体
長寿命のTT体の生成を抑制するためにピラン部位の4位の炭素とベンゼン部位の5位の炭素を架橋する方法がSousaらによって提案され、新しいタイプの縮環型ベンゾピランが合成された[49]。アセトニトリル溶液に室温で紫外光を照射しても溶液の色は無色のままで発色を目視することはできない。これは熱消色反応速度が大きいことを示唆しているが、レーザーフラッシュフォトリシス法により過渡吸収スペクトルを測定することで、発色体は300nmから650nmまでの幅広い波長領域に吸収帯を有し、全ての波長領域で単一成分の指数関数的減衰を示すことがわかった。すなわち、発色体はTC体の1種類のみであり、巧みな分子設計によりTT体の生成を抑制することに成功した。特筆すべきことは、発色体の室温での半減期はアセトニトリル溶液中で650マイクロ秒と従来のナフトピラン類と比較して大幅に高速化されたことである。
スピロピランの分子構造をもとにして着想されたナフトピラン類は、その優れた耐光性と高速熱消色特性からプラスチック調光レンズとして実用化されたが、調光材料以外の用途についてはほとんど検討されていないのが現状である。熱消色反応のさらなる高速化により、透過率、屈折率、蛍光などの光学特性に限らず、分子構造や双極子モーメントの大きな変化を利用した物質形状の高速光スイッチへの展開が期待される。
- オキサジン誘導体
スピロピランに紫外光を照射すると、数ピコ秒でC-O結合の開裂が起こり、引き続きマイクロ秒の時間領域でC=C結合周りのcis-trans熱異性化が起きることでメロシアニンを生成する[52]。発色体であるメロシアニンは、trans-cis熱異性化を起こした後に閉環反応によりスピロピランに戻るため、消色反応は比較的遅いものになる。例えば、スピロピランのニトロ誘導体である6-nitroBIPSでは、発色体の半減期は室温アセトニトリル溶液中で4分48秒である。Raymoらは、メロシアニンが有するC=C結合を生成しえない発色体を合理的に分子設計することで、熱消色反応の高速化を図った[53,54]。
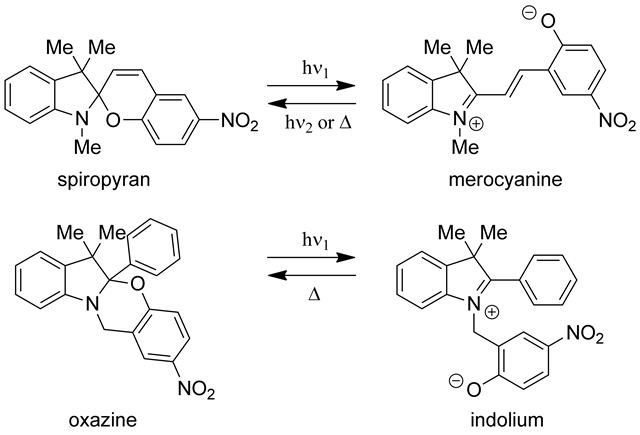
スピロピランとオキサジンのフォトクロミズム
無色の[1,3]オキサジンは紫外光を吸収することで、スピロピランと同様にC-O結合の開裂が起こり、p-ニトロフェノレート部位を有するインドリウムを生成する。ナノ秒レーザーフラッシュフォトリシス測定により、発色体であるインドリウムは440nmに吸収極大を持ち、室温アセトニトリル中の半減期が15ナノ秒という高速熱消色反応を示すことを報告した。スピロピラン類と比較すると熱消色反応の大幅な高速化が実現されている。また、インドリウムは独立したp-ニトロフェノレート部位を持つことに着目し、p-ニトロフェノレートアニオン分子のモル吸光係数を用いることで、紫外光照射により生成したインドリウムの分子数を推算してフォトクロミック反応の量子収率を約0.1と求めた[54]。さらに、パルス幅6ナノ秒、出力8mJのレーザーパルスを3,000回照射してもオキサジンの吸収スペクトルとインドリウムの過渡吸収スペクトルに変化が見られないことから、オキサジンが光反応に対して高い繰り返し耐久性を有していること、およびフォトクロミック反応は溶存酸素による影響を受けないことが示された。置換基がフォトクロミック反応に及ぼす影響についても詳細に検討されている[55,56]。興味深いことに、インドリウムを生成するオキサジンの開環反応は熱的にも進行し、熱開環反応の速度論的研究が1H NMRを用いて行われている[54]。すなわち、吸収スペクトルで確認することはできないが、室温下でも消色体のオキサジンと発色体のインドリウムの熱平衡混合物として存在していることがわかった。
オキサジンのように光反応量子収率が小さく、熱消色反応が速いと、通常の光照射条件下では目視による発色を確認することはできないので、調光レンズ材料としての用途には不適切な分子である。そこで、Raymoらはオキサジンの高速熱消色反応を利用して超解像蛍光顕微鏡用蛍光色素に供する蛍光高速光スイッチ分子に展開した[24-28]。通常の光学顕微鏡や蛍光顕微鏡では光の回折限界の問題があり、観測波長の半分程度以下の空間分解能を超えることはできないが、近年登場した超解像蛍光顕微鏡では回折限界をはるかに超える空間分解能を実現することが可能である[29]。Hellが考案したSTED(Stimulated
emission depletion)顕微鏡は、共焦点レーザー顕微鏡の光学系を用いて、蛍光観察用励起光とSTED光の2種類のレーザーをほぼ同時に試料に照射することで蛍光顕微鏡観察を行うものであり、測定原理的にはRESOLFT(Reversible
saturable optical fluorescence transitions)法の範疇に属する。STED光は励起状態をレーザーの発振原理になっている誘導放出により失活させるためのものであり、ビームの中心部が暗く、周辺部が明るいドーナッツ形状をしている。観察試料に対物レンズ側から中心位置を合わせた蛍光観察用励起光とSTED光を同時に照射すると、蛍光観察用励起光が照射されている部分から全方位に発せられる蛍光は対物レンズを通して観察することができるが、STED光が照射されている部分から発せられる誘導放出光はSTED光の進行方向に出射されるため対物レンズ側には届かない。すなわち、蛍光観察用励起光とSTED光が重なっている部分からの蛍光は観察されないことになる。蛍光観察用励起光のビーム径が400nm程度であっても、STED光のビームの中心部の暗い部分の径を50nmにすることができれば、蛍光観察用励起光の中心径50nmの部分からの蛍光のみを観察できることになり、光の回折限界を大きく超えることが可能となる。STED顕微鏡では、ドーナッツビームは誘導放出を起こすための役割を担っているが、フォトクロミック反応を誘起するために利用することもできる。すなわち、蛍光性フォトクロミック分子に紫外光照射することで生成する発色体が蛍光消光を起こす場合には、STED顕微鏡を利用して超解像蛍光顕微鏡像を観察することが可能になる。STED顕微鏡は蛍光観察用励起光とSTED光を同期して試料上を高速に走査することで、回折限界を超えた蛍光像を取得するので、近隣部位の蛍光観察を可能とするためには、蛍光消光状態から蛍光状態への迅速な回復が必要になる。すなわち、発色体から消色体への高速消色反応が要求されるが、P型フォトクロミック分子では消色反応の光反応量子収率が、T型フォトクロミック分子では熱消色反応速度が重要になる。
Raymoらは蛍光部位としてBODIPYを導入した蛍光性オキサジンを合成し、紫外光照射で生成したインドリウムでは、蛍光共鳴エネルギー移動(FRET:
Fluorescence resonance energy transfer)によりBODIPYの蛍光強度が8%減少することを報告した。蛍光強度の回復は熱消色反応速度と一致しており、マイクロ秒領域での蛍光強度変調が実現できた[25]。さらに、無蛍光性オキサジンに紫外光照射することで、蛍光性インドリウムを生成する高速熱消色型フォトクロミック分子についても報告しているが[26]、こちらの系は超解像蛍光顕微鏡の一つであるPALM(Photo-activated
localization microscopy)に応用することが可能である[29]。
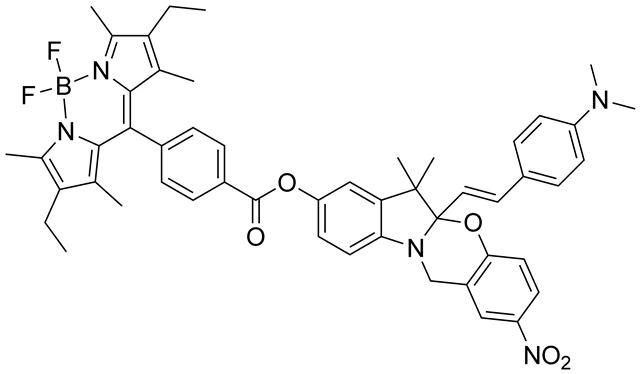
蛍光スイッチ機能を付与したオキサジン誘導体
このように、フォトクロミック分子を用いた蛍光強度のON−OFF光スイッチはSTEDやPALMのような光の回折限界を超えた超解像蛍光顕微鏡に応用できることから極めて重要な研究課題となっている。P型フォトクロミック分子を用いた蛍光スイッチは古くから研究されており実際に超解像蛍光顕微鏡観察に用いられているが[57-59]、蛍光のON−OFFを行うためには消色体を発色体に変換するための励起光源と、発色体を消色体に戻すための励起光源の二種類が必要になるのに対して、T型フォトクロミック分子では消色体を発色体に変換するための励起光源だけが必要になるところが特徴である。高速熱消色型フォトクロミック蛍光分子を超解像蛍光顕微鏡観察に用いるためには、発色反応の量子収率と蛍光強度のON/OFF比の改善が必要である。
- 参考文献
1) 柴田和雄、右衛門佐重雄、原富之、宮地重遠編、「光生物学(上)、(下)」、学会出版センター、東京(1979)
2) 古谷雅樹、「フィトクロム」、岩波書店、東京(1976)
3) N. Hampp, Chem. Rev., 100, 1755 (2000).
4) 神取秀樹、前田章夫、蛋白質核酸酵素、42、101 (1997)
5) 入江拓、佐賀佳央、渡辺正、生産研究、51、567 (1999)
6) 前田章夫、生化学、11、1005 (2008)
7) W. Kuhlbrandt, Nature, 406, 569 (2000).
8) A. Royant, K. Edman, T. Ursby, E. Pebay-peyroula, E. M. Landau and R. Neutze,
Nature, 406, 645 (2000).
9) H. J. Sass, G. Buldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J.
Berendzen and P. Ormos, Nature, 406, 649 (2000).
10) S. Subramaniam and R. Henderson, Nature, 406, 653 (2000).
11) 柴田幹大、生物物理、50、302 (2010)
12) M. Shibata, H. Yamashita, T. Uchihashi, H. Kandori and T. Ando, Nature Nanotech., 5, 208 (2010).
13) K. Sasaki and T. Nagamura, J. Appl. Phys., 83, 2894 (1998).
14) F. M. Raymo, Adv. Mater., 14, 401 (2002).
15) V. Balzani, A. Credi and M. Venturi, ChemPhysChem, 4, 49 (2003).
16) S. Giordani and F. M. Raymo, Org. Lett., 5, 3559 (2003).
17) M. Tomasulo, S. Giordani and F. M. Raymo, Adv. Funct. Mater., 15, 787 (2005).
18) M. Hofmann, C. Eggeling, S. Jakobs and S. W. Hell, Proc. Natl. Acad. Sci. USA, 102, 17565 (2005).
19) M. A. Schwentker, H. Bock, M. Hofmann, S. Jakobs, J. Bewersdorf, C. Eggeling
and S. W. Hell, Micros. Res. Tech., 70, 269 (2007).
20) S. W. Hell, Science, 316, 1153 (2007).
21) M. Heilemann, P. Dedecker, J. Hofkens and M. Sauer, Laser & Photon. Rev., 3, 180 (2009).
22) H. Mizuno, P. Dedecker, R. Ando, T. Fukano, J. Hofkens and A. Miyawaki,
Photochem. Photobiol. Sci., 9, 239 (2010).
23) P. C. Maurer, J. R. Maze, P. L. Stanwix, L. Jiang, A. V. Gorshkov, A. A. Zibrov, B. Harke, J. S. Hodges, A. S. Zibrov, A. Yacoby, D. Twitchen, S. W. Hell, R. L. Walsworth and M. D. Lukin, Nature Phys., 6, 912 (2010).
24) M. Tomasulo, E. Deniz, R. J. Alvarado and F. M. Raymo, J. Phys. Chem. C, 112, 8038 (2008).
25) E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 1, 1690 (2010).
26) E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 1, 3506 (2010).
27) E. Deniz, S. Ray, M. Tomasulo, S. Impellizzeri, S. Sortino and F. M. Raymo, J. Phys. Chem. A, 114, 11567 (2010).
28) E. Deniz, M. Tomasulo, J. Cusido, S. Sortino and F. M. Raymo, Langmuir, 27, 11773 (2011).
29) 堀田純一、水野秀昭、W. Sempels、J. Hofkens、光化学、42、121 (2011)
30) T. Yamamoto, M. Hasegawa, A. Kanazawa, T. Shiono and T. Ikeda, J. Mater. Chem., 10, 337 (2000).
31) S. Yoneyama, T. Yamamoto, O. Tsutsumi, A. Kanazawa, T. Shiono and T. Ikeda, Macromolecules, 35, 8751 (2002).
32) S. Tay, P.-A. Blanche, R. Voorakaranam, A. V. Tunc, W. Lin, S. Rokutanda,
T. Gu, D. Flores, P. Wang, G. Li, P. St Hilaire, J. Thomas, R. A. Norwood,
M. Yamamoto and N. Peyghambarian, Nature, 451, 694 (2008).
33) P.-A. Blanche, A. Bablumian, R. Voorakaranam, C. Christenson, W. Lin, T.
Gu, D. Flores, P. Wang, W.-Y. Hsieh, M. Kathaperumal, B. Rachwal, O. Siddiqui,
J. Thomas, R. A. Norwood, M. Yamamoto and N. Peyghambarian, Nature, 468, 80 (2010).
34) R. S. Becker and J. Michl, J. Am. Chem. Soc., 88, 5931 (1966).
35) J. Kolc and R. S. Becker, J. Phys. Chem., 71, 4045 (1967).
36) N. W. Tyer and R. S. Becker, J. Am. Chem. Soc., 92, 1289 (1970).
37) B. Van Gemert, Chapter 3, J. C. Crano and R. J. Guglielmetti, Organic Photochromic
and ThermochromicCompounds; Plenum Press: New York (1999).
38) S. Jockusch, N. J. Turro and F. R. Blackburn, J. Phys. Chem. A, 106, 9236 (2002).
39) W. Zhao and E. M. Carreira, Chem. Eur. J., 13, 2671 (2007).
40) N. Malic, J. A. Campbell and R. A. Evans, Macromolecules, 41, 1206 (2008).
41) F. Ercole, T. P. Davis and R. A. Evans, Macromolecules, 42, 1500 (2009).
42) F. Ercole, N. Malic, S. Harrisson, T. P. Davis and R. A. Evans, Macromolecules, 43, 249 (2010).
43) S. Han and Y. Chen, J. Mater. Chem., 21, 12402 (2011).
44) R. S. Becker, U. S. Pat., 3,567,605 (1971).
45) C. Lenoble and R. S. Becker, J. Photochem., 33, 187 (1986).
46) S. Delbare, J. Micheau and G. Vermeersch, J. Org. Chem., 68, 8968 (2003).
47) S. Delbaere and G. Vermeersch, J. Photochem. Photobiol. A: Chem., 159, 227 (2003).
48) P. J. Coelho, M. A. Salvador, M. M. Oliveira and L. M. Carvalho, J. Photochem. Photobiol. A: Chem., 172, 300 (2005).
49) C. M. Sousa, J. Pina, J. S. de Melo, J. Berthet, S. Delbaere and P. J.
Coelho, Org. Lett., 13, 4040 (2011).
50) B. Van Gemert, U. S. Pat., 5,645,767 (1997).
51) P. J. Coelho, M. A. Salvador, M. M. Oliveira and L. M. Carvalho, Tetrahedron, 60, 2593 (2004).
52) S. A. Krysanov and M. V. Alfimov, Chem. Phys. Lett., 91, 77 (1982).
53) M. Tomasulo, S. Sortino and F. Raymo, Org. Lett., 7, 1109 (2005).
54) M. Tomasulo, S. Sortino, A. J. P. White and F. Raymo, J. Org. Chem., 70, 8180 (2005).
55) E. Deniz, M. Tomasulo, S. Sortino and F. Raymo, J. Phys. Chem. C., 113, 8491 (2009).
56) M. Tomasulo, S. Sortino and F. Raymo, J. Org. Chem., 73, 118 (2008).
57) R. Ando, H. Mizuno and A. Miyawaki, Science, 306, 1370 (2004).
58) K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 133, 13558 (2011).
59) K. I. Willig, A. C. Stiel, T. Brakemann, S. Jakobs and S. W. Hell, Nano Lett., 11, 3970 (2011).